SQUID | Cracking Open the Black Box of Genomic DNNs

AI is capable of trawling through unfathomable quantities of genetic data points to pick out novel therapeutic targets. However, little is understood about how the ‘black box’ algorithms actually achieve such results. A new computational tool called SQUID (Surrogate Quantitative Interpretability for Deepnets) is attempting to address this.
The inner workings of deep neural networks (DNNs) are complicated beyond human comprehension. They rely on millions of intricately tuned connections between artificial neurons that transform input data as it passes through many layers.
The complexity of these connections is a double-edged sword. It facilitates an unparalleled ability to detect intricate patterns in data, but also means it is extremely challenging to understand how DNNs work.
This opacity can be particularly troubling in fields like genomics, where understanding the rationale behind predictions is crucial for advancing scientific knowledge and clinical applications. Without clarity on how AI models draw their conclusions, scientists face difficulties in validating results and applying them to real-world scenarios.
Genomic DNNs excel in predicting a wide array of genomic activities, such as mRNA expression, protein-DNA binding, and chromatin accessibility. However, extracting mechanistic insights from these models is challenging due to the nonlinearities and noise present in functional genomics data.
Traditional attribution methods, such as Saliency Maps and DeepLIFT, often provide inconsistent motifs across different genomic sequences, leading to varied and sometimes unreliable biological interpretations.
Developed by researchers at Cold Spring Harbor Labratory (CSHL; NY, USA) SQUID addresses these limitations by using surrogate models to approximate DNNs in specific regions of sequence space.
Surrogate models are simpler and mechanistically interpretable, allowing for clearer insights into the biological mechanisms underlying DNN predictions. The SQUID framework involves three main steps:
- Generate an In Silico MAVE Dataset: This involves creating a library of variant sequences and using the DNN to assign functional scores to each sequence.
- Fit a Surrogate Model: The surrogate model, which has interpretable parameters, is trained on the in silico data.
- Visualize and Interpret the Surrogate Model: The parameters of the surrogate model are visualized to uncover biological mechanisms.
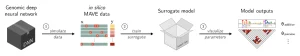
SQUID computational pipeline. Credit: Cold Spring Harbor Labratory
It allows scientists to conduct thousands of virtual experiments simultaneously, effectively “fishing out” the most accurate algorithms behind the AI’s predictions. Such computational “catches” can pave the way for more realistic and grounded laboratory experiments.
When benchmarked against established attribution methods on various genomic DNNs, SQUID was found to significantly reduce non-biological noise in attribution maps.
This noise reduction is crucial for accurately identifying weak transcription factor binding sites, which play vital roles in gene regulation but are often difficult to detect due to their subtle signals.
SQUID’s improved noise management also enhances the prediction of single-nucleotide variant effects, providing better insights into which genetic variants might be pathogenic.
Assistant Professor Peter Koo of CSHL highlights a critical aspect of SQUID’s superior performance: its specialized training. Traditional tools for interpreting AI models often stem from fields like computer vision or natural language processing. While these methods have their merits, they fall short in genomics. Koo elaborates:
“The tools that people use to try to understand these models have been largely coming from other fields like computer vision or natural language processing. While they can be useful, they’re not optimal for genomics. What we did with SQUID was leverage decades of quantitative genetics knowledge to help us understand what these deep neural networks are learning.”
By providing clearer insights into AI models’ decision-making processes, it helps bridge the gap between computational predictions and real-world biological research, potentially leading to groundbreaking discoveries in the field of genetics.
This research also follows recent attempts by OpenAI to demystify neural activity within their GPT-4 model and Anthropic to reverse engineer Claude.